Neurodegenerative Erkrankungen stellen eine der größten Herausforderungen unserer alternden Gesellschaft dar. Allerdings lassen sie sich im Labor nur schwer erforschen. Geeignete Tiermodelle sind rar und menschliches Gehirngewebe ist nur eingeschränkt verfügbar. Wissenschaftler des Nationalen Genomforschungsnetzes NGFN haben nun zur Erforschung einer erblichen Bewegungsstörung einen Umweg genommen: Sie wandelten Hautzellen von Patienten mit der Machado-Joseph-Erkrankung in sogenannte induzierte pluripotente Stammzellen um und gewannen daraus funktionierende Nervenzellen. (Newsletter 56 / März 2012)
![]() An diesen künstlich hergestellten Nervenzellen entschlüsselten sie die Ursache der Krankheit. Erstmals wurde die Erkrankung bei portugiesischstämmigen Bewohnern der Azoren beschrieben. In Deutschland ist sie heute die häufigste dominant vererbte Bewegungsstörung: die Machado-Joseph Erkrankung, auch spinocerebelläre Ataxie 3 genannt. Die Mehrzahl der Betroffenen entwickelt zwischen dem 20. und 40. Lebensjahr Gangstörungen und eine Reihe weiterer neurologischer Symptome. Die Ursache der Erkrankung liegt in den Genen: Das Ataxin-3-Gen enthält einen Abschnitt, der bei Patientinnen und Patienten mit der Machado-Joseph-Erkrankung in zahlreichen Wiederholungen im Genom vorliegt. Die Folge: Das entsprechende Protein wird zwar gebildet, verklumpt aber, wodurch schließlich die Nervenzellen im Gehirn geschädigt werden. Unklar war bislang, warum die Erkrankung nur Nervenzellen betrifft und wie die abnorme Proteinverklumpung ausgelöst wird.
An diesen künstlich hergestellten Nervenzellen entschlüsselten sie die Ursache der Krankheit. Erstmals wurde die Erkrankung bei portugiesischstämmigen Bewohnern der Azoren beschrieben. In Deutschland ist sie heute die häufigste dominant vererbte Bewegungsstörung: die Machado-Joseph Erkrankung, auch spinocerebelläre Ataxie 3 genannt. Die Mehrzahl der Betroffenen entwickelt zwischen dem 20. und 40. Lebensjahr Gangstörungen und eine Reihe weiterer neurologischer Symptome. Die Ursache der Erkrankung liegt in den Genen: Das Ataxin-3-Gen enthält einen Abschnitt, der bei Patientinnen und Patienten mit der Machado-Joseph-Erkrankung in zahlreichen Wiederholungen im Genom vorliegt. Die Folge: Das entsprechende Protein wird zwar gebildet, verklumpt aber, wodurch schließlich die Nervenzellen im Gehirn geschädigt werden. Unklar war bislang, warum die Erkrankung nur Nervenzellen betrifft und wie die abnorme Proteinverklumpung ausgelöst wird.
 Um den Krankheitsprozess auf molekularer Ebene zu studieren, stellten Wissenschaftler um den Stammzellforscher Prof. Dr. Oliver Brüstle vom Institut für Rekonstruktive Neurobiologie der Universität Bonn zunächst aus kleinen Hautproben von Patienten sogenannte induzierte pluripotente Stammzellen, kurz iPS-Zellen, her. „Es handelt sich dabei um Zellen, die in ein sehr frühes, undifferenziertes Stadium zurückversetzt werden“, erklärt Professor Brüstle. Diese „Alleskönner“ lassen sich – einmal gewonnen – nahezu uneingeschränkt vermehren und in alle Körperzellen ausreifen. In einem nächsten Schritt wandelten die Wissenschaftler die iPS-Zellen in Gehirnstammzellen um, aus denen sie beliebig Nervenzellen für ihre Untersuchungen entwickeln konnten.
Um den Krankheitsprozess auf molekularer Ebene zu studieren, stellten Wissenschaftler um den Stammzellforscher Prof. Dr. Oliver Brüstle vom Institut für Rekonstruktive Neurobiologie der Universität Bonn zunächst aus kleinen Hautproben von Patienten sogenannte induzierte pluripotente Stammzellen, kurz iPS-Zellen, her. „Es handelt sich dabei um Zellen, die in ein sehr frühes, undifferenziertes Stadium zurückversetzt werden“, erklärt Professor Brüstle. Diese „Alleskönner“ lassen sich – einmal gewonnen – nahezu uneingeschränkt vermehren und in alle Körperzellen ausreifen. In einem nächsten Schritt wandelten die Wissenschaftler die iPS-Zellen in Gehirnstammzellen um, aus denen sie beliebig Nervenzellen für ihre Untersuchungen entwickeln konnten.
Das Besondere: Da die Nervenzellen aus den Patienten selbst stammen, tragen sie dieselben genetischen Veränderungen und können so als zelluläres Modell der Erkrankung dienen. „Diese Methode erlaubt uns die Erforschung der Erkrankung an den wirklich betroffenen Zellen, zu denen wir sonst keinen Zugang hätten – fast so, als hätten wir das Gehirn des Patienten in die Zellkulturschale gebracht“, sagt Dr. Philipp Koch, langjähriger Mitarbeiter von Professor Brüstle.
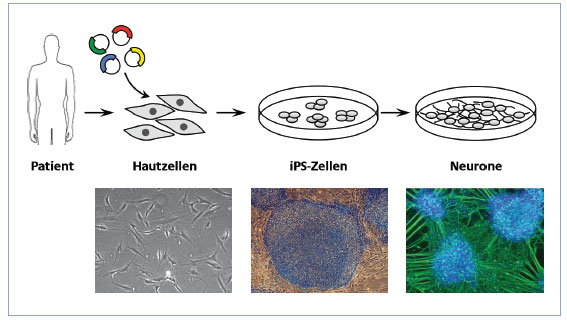 Hautzellen von Patienten (links) werden mit Hilfe von verschiedenen Faktoren zu iPS-Zellen (Mitte) umprogrammiert und dann weiter zu Nervenzellen (rechts) ausgereift.
Hautzellen von Patienten (links) werden mit Hilfe von verschiedenen Faktoren zu iPS-Zellen (Mitte) umprogrammiert und dann weiter zu Nervenzellen (rechts) ausgereift.
Die Forschungsarbeiten wurden vom Bundesministerium für Bildung und Forschung (BMBF) im Rahmen des Verbunds DiGtoP („From Disease Genes to Proteins“, Koordinator Prof. Dr. Wolfgang Wurst) im Nationalen Genomforschungsnetz NGFN-Plus unterstützt.
Das Team um Professor Brüstle stimulierte die künstlich geschaffenen Nervenzellen elektrisch. Dabei konnten die Forscherinnen und Forscher zeigen, dass die Bildung der Proteinaggregate unmittelbar mit der elektrischen Aktivität der Nervenzellen zusammenhängt. „Eine Schlüsselrolle spielt dabei das Enzym Calpain, das durch den erhöhten Kalziumgehalt stimulierter Nervenzellen aktiviert wird“, so Dr. Peter Breuer von der Klinik und Poliklinik für Neurologie des Bonner Universitätsklinikums. „Dieser neu identifizierte Mechanismus erklärt, warum die Erkrankung ausschließlich Nervenzellen betrifft“, betont Professor Brüstle.
In einem nächsten Schritt wollen Professor Brüstle und seine Kollegen reprogrammierte Nervenzellen für die Entwicklung von Wirkstoffen zur Behandlung neurologischer Erkrankungen einsetzen.
